Pulmonary Fibrosis Life Expectancy: Prognosis
Upon diagnosis, pulmonary fibrosis life expectancy can vary depending on many different factors. Pulmonary fibrosis (PF), or scarring of the lungs, consists of over 200 types of lung ailments that are difficult to distinguish. All of them fall into a larger family of lung conditions, called interstitial lung disease (ILD), which involves inflammation and/or scarring of the lung tissue. The most common type of pulmonary fibrosis is idiopathic pulmonary fibrosis (IPF), which is when lung scarring occurs with no known cause—a condition that affects one in 200 U.S. adults over the age of 70.
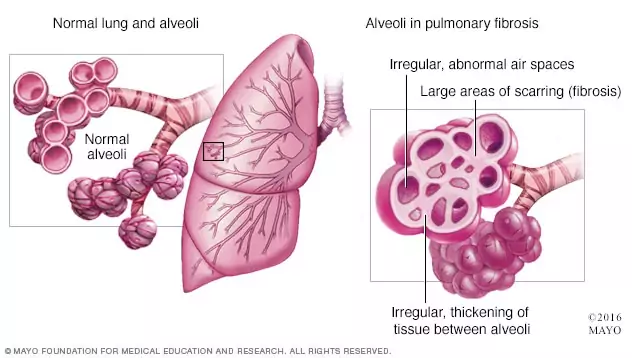
This image is from the Mayo Clinic.
All types of PF involve progressive scarring and inflammation. As scarring builds, so does breathlessness, and lung failure can eventually develop. The Pulmonary Fibrosis Foundation (PFF) notes there is no way to foresee how long someone with PF or IPF will live. The average life expectancy of pulmonary fibrosis is said to be three to five years after diagnosis, but those figures are now outdated.
Disease progression can be impacted, both positively and negatively, by a number of different factors. The Pulmonary Fibrosis Foundation (PFF) and the University of Rochester Medical Center point out several factors that have a major influence on a PF patient’s prognosis.
Factors that impact pulmonary fibrosis life expectancy
Causes linked to a positive prognosis:
- Early detection: Early detection plays a large role in a patient’s prognosis. The sooner PF or IPF is diagnosed, the better one’s chance of survival. Disease severity at the time of diagnosis is directly related to one’s life expectancy.
- Anti-scarring treatments: While there is no cure for pulmonary fibrosis, two anti-fibrotic drugs, or anti-scarring treatments, have been approved by the U.S. Food and Drug Administration (FDA) and have clinically proven to slow PF progression.
- Individual differences: Lung function may decline faster in some patients than others. While many live past three to five years, others experience respiratory failure before the three-year mark, and some become very ill within months.
Causes linked to a negative prognosis
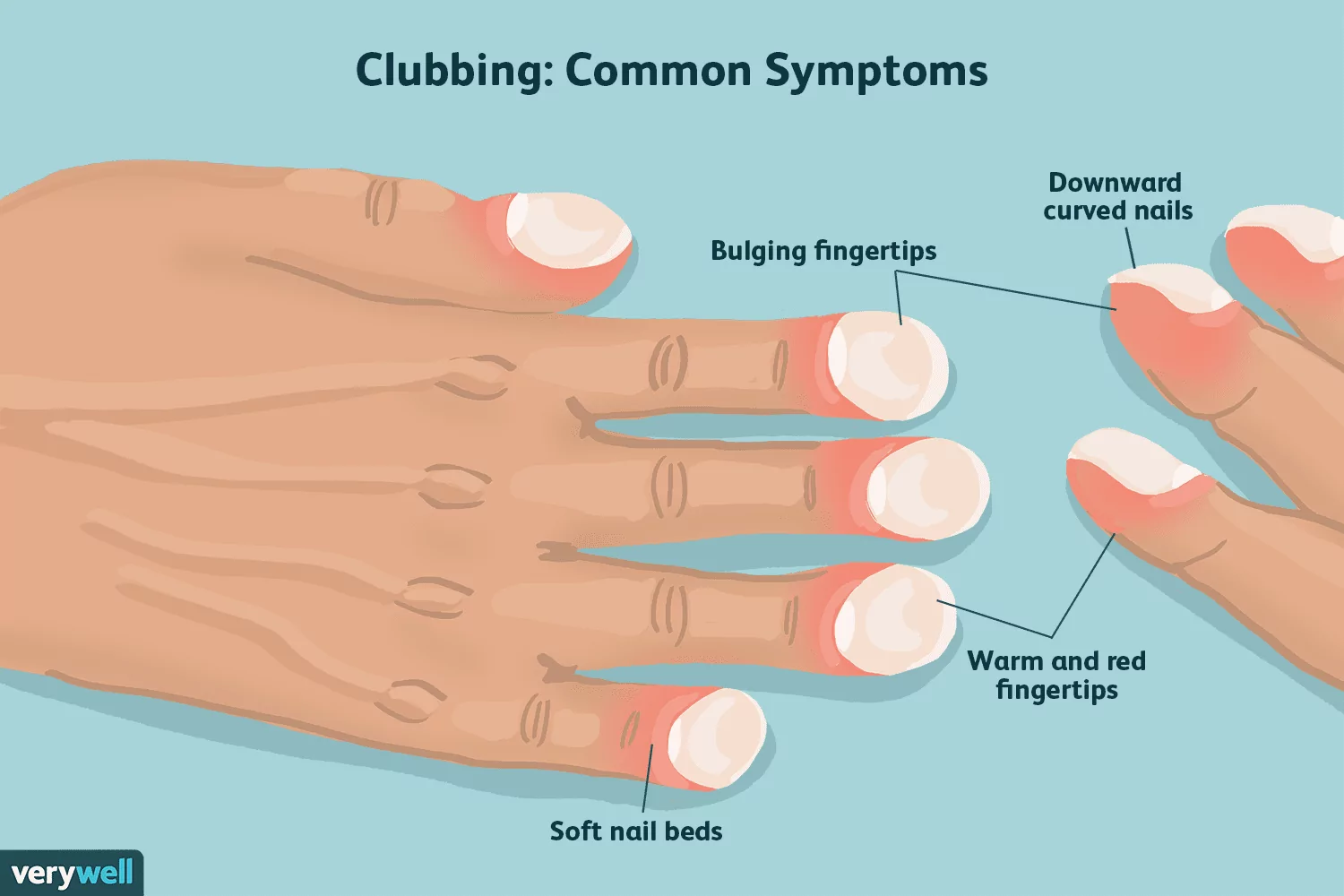
This image is from Verywell Health.
A number of clinical factors are associated with worse prognosis, or reduced survival rate, with IPF:
- While the median age (age range midpoint) of an IPF diagnosis is 66 years old, older age is linked to worse survival rates. One study showed those under the age of 50 had a median survival of 9.7 years; those aged 50-60 had roughly 5.2 years; and those aged 60-70 had about 2.3 years.
- In general, current and former smokers have a worse survival rate than nonsmokers with IPF.
- Increased breathlessness, or dyspnea, has proven to be an important predictor of survival after accounting for disease severity and other disease factors. Dyspnea is measured by changing scores in two measures for IPF, such as the modified Medical Research Council (mMRC).
- IPF patients with a lower BMI (body mass index) have shorter lives than those with higher BMIs. The above study showed a median lifespan of 3.6 years for a BMI of under 25; 3.8 years for a BMI of 25-30; and 5.8 years for a BMI of over 30. This protective effect of a low BMI may signal malnutrition or a greater caloric expenditure.
- After accounting for age and smoking history, the study also showed that clubbed fingers are strongly linked to a shorter life expectancy from pulmonary fibrosis, though the American Thoracic Society (ATC) says further studies are needed.
- Declining lung function is linked to a worse prognosis and seen as a decrease in forced vital capacity (FVC), total lung capacity (TLC), and diffusing capacity of the lungs for carbon monoxide (DLCO).
- Greater fibrosis on a high-resolution computed tomography (HRCT) scan signals IPF progression, with a UIP pattern (usual interstitial pneumonia) generally linked to worse life expectancy.
- Concurrent diseases, like pulmonary hypertension and gastroesophageal reflux, can make PF worse and lower one’s survival rate.
Additional reading on pulmonary fibrosis life expectancy can be found on Pulmonary Fibrosis News and the American Lung Association.